UniPTM: Multiple PTM site prediction on full-length protein sequence
Abstract
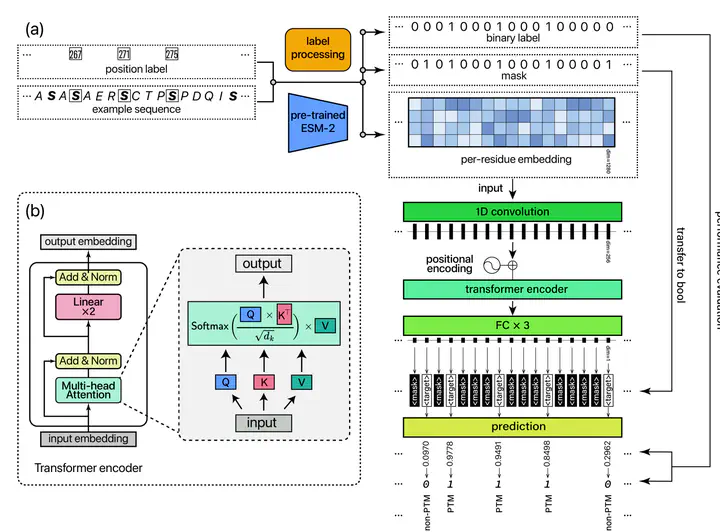
Post-translational modifications (PTMs) enrich the functional diversity of proteins by attaching chemical groups to the side chains of amino acids. In recent years, a myriad of AI models have been proposed to predict many specific types of PTMs. However, those models typically adopt the sliding window approach to extract short and equal-length protein fragments from full-length proteins for model training. Unfortunately, such a subtle step results in the loss of long-range information from distal amino acids, which may impact the PTM formation process. In this study, we introduce UniPTM, a window-free model designed to train and test on natural and full-length protein sequences, enabling the prediction of multiple types of PTMs in a holistic manner. Moreover, we established PTMseq, the first comprehensive dataset of full-length protein sequences with annotated PTMs, to train and validate our model. UniPTM has undergone extensive validations and significantly outperforms existing models, elucidating the influence of protein sequence completeness on PTM. Consequently, UniPTM offers interpretable and biologically meaningful predictions, enhancing our understanding of protein functionally and regulation. The source code and PTMseq dataset for UniPTM are available at https://uniptm.com.
Highlights:
1. UniPTM takes full-length protein sequence data as input, considering all amino acids across the entire protein that could influence the occurrence of PTMs, even if they are distal.
2. PTMseq dataset consists subsets of nine types of PTMed sequences, currently encompassing a total of 34,514 distinct PTM sites annotated across 12,203 full-length protein sequences.
3. The pre-trained ESM-2 model embeds contextual information from the entire sequence into each residue, allowing every target amino acid to engage independently.
Supplementary Information can be accessed here.
Ke Cheng
Postdoctoral Researcher
My research interests lie at the surface of chemistry and biology, where I am deeply passionate about applying innovative chemistry to advance fields such as chemoproteomics, drug discovery, nanomedicine, and theranostics. My aim is to provide robust methodologies for mapping biological interactomes, accelerating drug development, and expanding therapeutic opportunities.