TransPTM: a transformer-based model for non-histone acetylation site prediction
 Copyright © 2024 Oxford University Press
Copyright © 2024 Oxford University PressAbstract
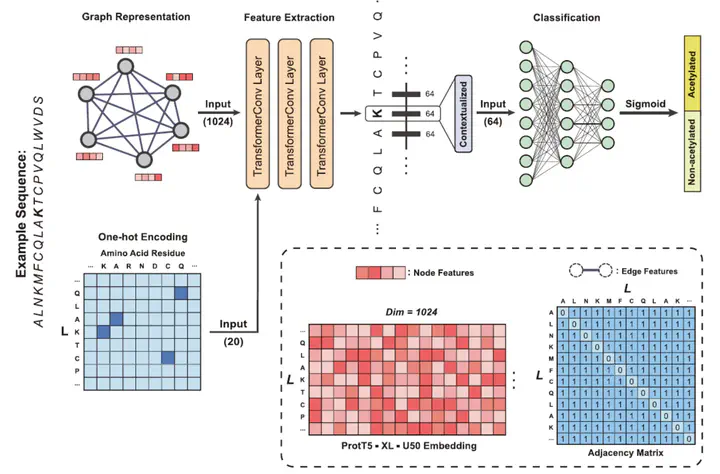
Protein acetylation is one of the extensively studied post-translational modifications (PTMs) due to its significant roles across a myriad of biological processes. Although many computational tools for acetylation site identification have been developed, there is a lack of benchmark dataset and bespoke predictors for non-histone acetylation site prediction. To address these problems, we have contributed to both dataset creation and predictor benchmark in this study. First, we construct a non-histone acetylation site benchmark dataset, namely NHAC, which includes 11 subsets according to the sequence length ranging from 11 to 61 amino acids. There are totally 886 positive samples and 4707 negative samples for each sequence length. Secondly, we propose TransPTM, a transformer-based neural network model for non-histone acetylation site predication. During the data representation phase, per-residue contextualized embeddings are extracted using ProtT5 (an existing pre-trained protein language model). This is followed by the implementation of a graph neural network framework, which consists of three TransformerConv layers for feature extraction and a multilayer perceptron module for classification. The benchmark results reflect that TransPTM has the competitive performance for non-histone acetylation site prediction over three state-of-the-art tools. It improves our comprehension on the PTM mechanism and provides a theoretical basis for developing drug targets for diseases. Moreover, the created PTM datasets fills the gap in non-histone acetylation site datasets and is beneficial to the related communities. The related source code and data utilized by TransPTM are accessible at https://www.github.com/TransPTM/TransPTM.
Highlights:
1. A transformer-based deep learning model is proposed for non-histone acetylation site prediction, which achieves the best performance among the state-of-the-art models.
2. The establishment of NHAC, a benchmark dataset specifically designed for non-histone acetylation site prediction, enriches the landscape of lysine acetylation site databases.
3. Pre-trained protein language model ProtT5 is employed for generating contextualized representations of protein sequences by capturing dependencies between amino acid residues.
4. The self-attention mechanism in TransPTM can reveal key residue positions for non-histone protein acetylation, thereby demonstrating the interpretability of our model.
Supplementary Information can be accessed here.
Ke Cheng
Postdoctoral Researcher
My research interests lie at the surface of chemistry and biology, where I am deeply passionate about applying innovative chemistry to advance fields such as chemoproteomics, drug discovery, nanomedicine, and theranostics. My aim is to provide robust methodologies for mapping biological interactomes, accelerating drug development, and expanding therapeutic opportunities.